How To Become CE Certified For Medical Devices In Europe: A No-Nonsense Guide
So, you’ve got a brilliant medical device. Something that could genuinely change lives, right? Great. Now comes the paperwork mountain. Specifically, getting that coveted CE mark for medical devices in Europe. It’s not for the faint of heart, and honestly, a lot of the official guidance feels like it was written by someone who hates sunshine and clarity.
Forget the jargon. We’re talking about making sure your product is safe, effective, and legal to sell across the pond. This isn’t just a rubber stamp; it’s a serious commitment to quality and patient safety. Let’s break down this beast, step-by-step, so you know exactly what you’re up against and how to conquer it.
Understanding the CE Marking Framework

Source : scilife.io
First off, what even is the CE mark? It’s not about European origin; it’s about conformity. Your device meets the stringent European health, safety, and environmental protection requirements. Think of it as your device’s passport to the European Economic Area (EEA).
What the CE Mark Signifies
This isn’t just a badge of honor. It means your device has gone through the necessary hoops to prove it’s safe and works as intended. For manufacturers, it signifies adherence to the Medical Device Regulation (MDR) or the In Vitro Diagnostic Regulation (IVDR), depending on your product. It’s a critical signal to authorities and end-users alike.
The Regulatory Bodies Involved
You’ll be dealing with a few key players. National competent authorities oversee the market, but it’s often the Notified Bodies that do the heavy lifting. These are independent organizations designated by EU countries to assess your device’s conformity. They’re the gatekeepers, essentially.
MDR vs. IVDR: Know Your Alphabet Soup
It’s Key to get this right. The MDR covers most medical devices, while the IVDR specifically targets in vitro diagnostic devices. Mixing them up? That’s a fast track to delays and headaches. Make sure you know which regulation applies to your specific product.
Source : operonstrategist.com
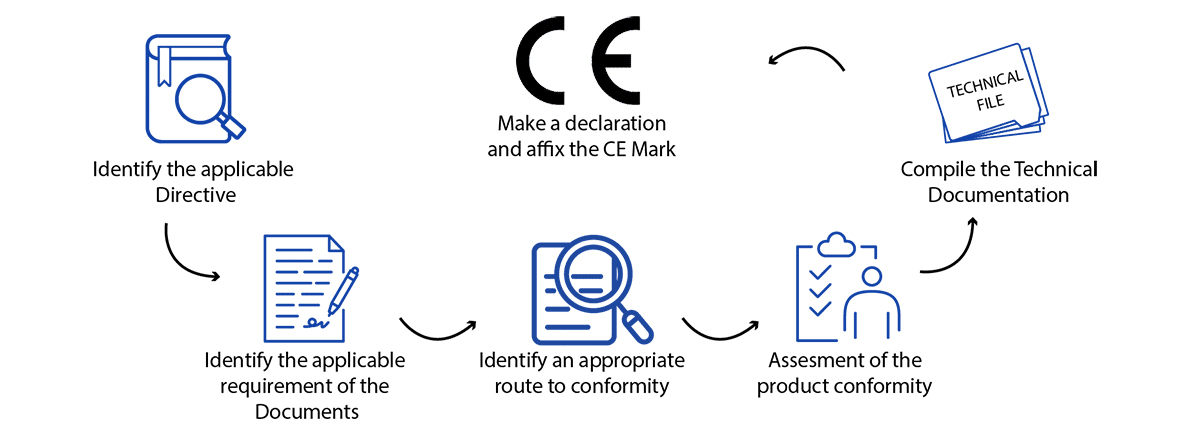
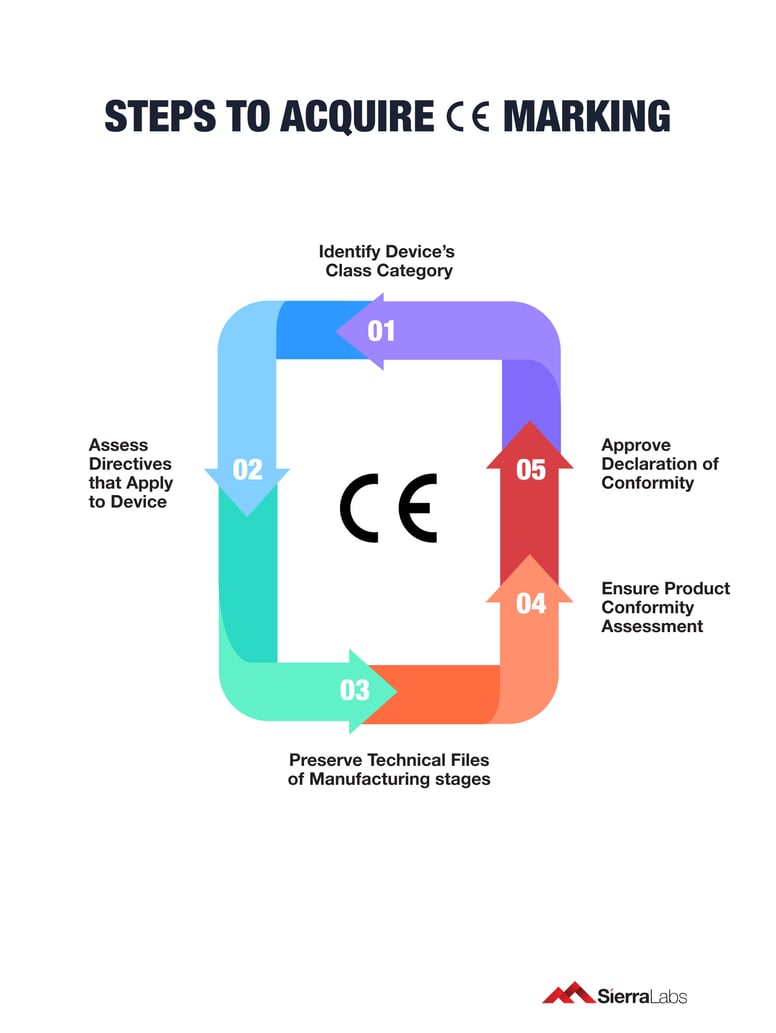
Device Classification: The First Key Step
This is where it all begins. How does the EU classify your medical device? It’s not arbitrary; it’s based on risk. The higher the risk your device poses to patients, the stricter the requirements will be.
Risk-Based Classification System
Devices are generally classified into Class I, Class IIa, Class IIb, and Class III for MDR, with Class III being the highest risk. For IVDs, it’s Class A, B, C, and D. This classification dictates much of the subsequent conformity assessment path. So, get this wrong, and everything else is built on shaky ground.
How to Determine Your Device’s Class
It involves carefully reviewing the classification rules laid out in the MDR or IVDR annexes. You need to consider the device’s intended purpose, invasiveness, duration of use, and whether it involves certain technologies or biological substances. Don’t guess here; consult the regulations meticulously or seek expert advice.
Implications of Different Classes
A Class I device (lowest risk) might have a relatively straightforward path, potentially even self-declaration of conformity. Higher classes (like Class III) demand significant scrutiny, usually involving a Notified Body assessment of your technical documentation and quality management system. It’s a sliding scale of complexity.
Quality Management System (QMS): The Backbone of Compliance
You absolutely cannot get a CE mark without a Strong Quality Management System. This isn’t just about ticking boxes; it’s about embedding quality and safety into your company’s DNA.
ISO 13485: The Gold Standard
Most manufacturers aim for certification under ISO 13485, the international standard for medical device QMS. It harmonizes with the MDR/IVDR requirements and provides a framework for consistently producing safe and effective devices. It covers everything from design and development to production and post-market surveillance.
Implementing Your QMS
Setting up a QMS means documenting your processes: design controls, risk management, supplier controls, document control, production, quality control, complaint handling, and more. It needs to be practical and followed religiously by everyone in the organization. It’s your operational bible.
QMS Audit by a Notified Body
For most device classes, your QMS will be audited by a Notified Body as part of the conformity assessment. They’ll want to see proof that your system is not just documented but actively implemented and effective. Be prepared for detailed inspections.
Technical Documentation: Your Device’s Blueprint
This is the Full dossier demonstrating your device’s safety and performance. Think of it as the ultimate sales pitch to the regulatory authorities.
What Goes into the Technical Documentation?
Source : blog.scalehealth.com
It’s a big file! It includes device descriptions, specifications, manufacturing information, labeling, and crucially, the results of your design verification and validation activities. You’ll also need to detail your risk management process and outcomes.
Clinical Evaluation: Proving Efficacy and Safety
This is a Foundation. You need to systematically gather and analyze clinical data to demonstrate that your device is safe and performs as intended. This might involve literature reviews, clinical investigations, or equivalence data. The depth required is significant, especially for higher-risk devices.
The MDR places a strong emphasis on clinical evidence. It’s not enough to just say it works; you need solid proof. This process is ongoing, too; it feeds into your post-market surveillance.
The Role of a Clinical Evaluation Report (CER)
The Clinical Evaluation Report (CER) is a key part of your technical documentation. It synthesizes all the clinical data, assesses its sufficiency, and concludes on the device’s safety and performance. This report must be updated throughout the device’s lifecycle.
Conformity Assessment Routes: Choosing Your Path
Once you have your QMS and technical documentation in order, you need to select the right conformity assessment route. The MDR/IVDR outline several options, largely dependent on your device’s classification.
Common Assessment Routes Explained
For lower-risk devices (like some Class I), you might only need to perform a self-assessment and issue a Declaration of Conformity. However, for most devices, especially medium and high-risk ones, involvement of a Notified Body is mandatory. This often involves auditing your QMS and reviewing your technical documentation.
Notified Body Involvement: When and Why
If your device falls into Class IIa, IIb, or III (or Class B, C, D for IVDs), a Notified Body must be involved. They provide the independent assurance that your device meets the Needed requirements. Without their stamp of approval (via an audit certificate), you can’t legally affix the CE mark.
Choosing the right Notified Body is also critical. Make sure they are designated for your specific device type and classification. The process of engaging with them can be lengthy, so start early.
The EU Declaration of Conformity
This is the manufacturer’s legally binding document stating that the device complies with all applicable EU regulations. Once you’ve successfully navigated the chosen conformity assessment route, you can draw this up. It’s the final sign-off before you add the CE mark.
Appointing an EU Authorized Representative
If you’re based outside the EU, this is non-negotiable. You legally need an EU Authorized Representative (A.R.). They act as your link to the EU authorities.
Who Can Be an Authorized Representative?
Watch: Process to Get CE Certification for Medical Devices | CE …
An A.R. must be a legally established entity within the EU. They are responsible for specific tasks, including verifying your Declaration of Conformity and technical documentation, and cooperating with competent authorities. Their role is Key for non-EU manufacturers.
Responsibilities of the Authorized Representative
Think of them as your local point of contact and accountability partner. They Make sure you’ve met your obligations and can be contacted by authorities if issues arise. It’s Key to have a clear contract outlining responsibilities.
For non-European manufacturers, Handling this requirement is Needed for market access. Companies specializing in regulatory affairs often offer these services, providing a Key bridge to the European market. This is particularly relevant for those unfamiliar with the intricacies of EU medical device regulations.
The Notified Body Audit Process
This is often the most daunting phase. It’s where an independent third party scrutinizes your QMS and technical documentation.
Preparing for the Audit
Thorough preparation is key. Make sure all your QMS documentation is up-to-date, accessible, and consistently applied. Have your technical files meticulously organized and ready for review. Simulate the audit yourself beforehand.
What the Auditors Look For
They’ll be checking for compliance with the MDR/IVDR, ISO 13485, and any specific requirements related to your device class. They examine your design controls, risk management, clinical evaluation, manufacturing processes, and post-market surveillance plans. Expect them to dig deep.
Post-Audit Actions and Certificates
After the audit, you’ll receive a report. If there are findings, you’ll need to address them through corrective actions. Once satisfied, the Notified Body will issue your CE certificate, a critical document for your market access.
Post-Market Surveillance and Vigilance
Getting the CE mark isn’t the finish line; it’s the starting pistol for ongoing monitoring.
The Importance of PMS
The EU MDR and IVDR place a massive emphasis on Post-Market Surveillance (PMS). You must proactively collect and analyze data about your device’s performance and safety once it’s on the market. This is about continuous improvement and patient safety.
Reporting Adverse Events
Serious incidents must be reported to the relevant authorities promptly. This vigilance system ensures that potential risks are identified and managed quickly across the EU. It’s a critical feedback loop.

Source : youtube.com
Updating Technical Documentation and CER
Your technical documentation, especially the clinical evaluation, isn’t static. PMS data feeds back into it, requiring updates to Make sure it always reflects the current state of knowledge and device performance. This continuous lifecycle approach is mandated.
Affixing the CE Mark and Market Entry
You’ve done the hard yards. Now for the final steps.
How to Apply the CE Mark
Once you have your EU Declaration of Conformity and, if required, your Notified Body certificate, you can affix the CE mark to your device, its packaging, or the instructions for use, as specified in the regulations. Make sure it’s visible and legible.
Device Registration (EUDAMED)
Depending on the device type and your location, you may need to register your device in the European database for medical devices (EUDAMED). This centralizes information and enhances transparency across the EEA. Full implementation is ongoing but Key.
Your Device is Ready for the EU Market!
Congratulations! With the CE mark affixed and all registration requirements met, your medical device is now legally compliant and ready to be sold throughout the European Economic Area. This opens up a vast market for your innovative products.
Common Pitfalls and How to Avoid Them
Plenty of manufacturers stumble. Knowing the common traps can save you immense pain.
Misclassifying Your Device
Getting the classification wrong is a fundamental error that cascades. Always err on the side of caution and consult experts if unsure. The consequences of a wrong classification can be severe, leading to significant rework.
Inadequate Clinical Evidence
Underestimating the rigor of clinical evaluation is a major mistake. The MDR demands Strong data. Insufficient clinical data is a frequent reason for Notified Body rejection.
Poorly Implemented QMS
A QMS that exists only on paper, without real-world application, will be spotted during an audit. You need a living, breathing system that your entire team follows. Proving implementation is as important as having the procedures.
Ignoring Post-Market Obligations

Source : decomplix.com
Thinking the work is done after certification is a dangerous assumption. The EU regulators are increasingly focused on post-market performance and safety. Failing here can lead to recalls and loss of CE marking. Active post-market surveillance is not optional.
Handling the European Maze: Expert Help is Available
Let’s be blunt: this process is complex. Trying to go it alone can be a recipe for disaster, especially for smaller companies or those new to the European market. Engaging regulatory consultants can streamline the process immensely.
When to Bring in the Experts
If you’re unsure about classification, QMS requirements, technical documentation, or the audit process itself, professional help is Crucial. It can save you time, money, and prevent costly mistakes.
Services Regulatory Consultants Offer
They can assist with everything from initial strategy and device classification to QMS implementation, technical file preparation, Notified Body liaison, and ongoing compliance. For non-European manufacturers, finding a reliable EU Authorized Representative is also a key service they provide. Companies often need help understanding the nuances of the European medical devices regulation (MDR) CE marking process.
Choosing the Right Partner
Look for consultants with a proven track record specifically in EU medical device regulations. Ask for references and understand their approach. A good partner won’t just tell you what to do; they’ll help you build the capability within your organization. Resources like CMC Medical Devices offer Full guides for non-European manufacturers.
Achieving CE certification for medical devices in Europe is a rigorous but achievable goal. It demands Careful planning, Strong systems, and a commitment to safety. By understanding the key requirements and potential challenges, you can successfully navigate the regulatory Field and bring your innovative medical solutions to the European market.
| Stage | Description | Key Considerations | Typical Involvement |
|---|---|---|---|
| 1. Classification | Determine the risk class of your device (I, IIa, IIb, III or A, B, C, D for IVDs). | Intended purpose, invasiveness, duration, materials. | Manufacturer (with potential expert review) |
| 2. QMS Implementation | Establish and document a Quality Management System compliant with ISO 13485. | Covers design, production, risk management, post-market surveillance. | Manufacturer (internal process) |
| 3. Technical Documentation | Compile Full device data proving safety and performance. | Device specs, risk analysis, clinical evaluation, manufacturing info. Getting CE certification requires detailed documentation. | Manufacturer |
| 4. Clinical Evaluation | Gather and analyze clinical data to demonstrate safety and efficacy. | Literature review, clinical investigations, equivalence. Key for MDR compliance. | Manufacturer (often supported by clinical experts) |
| 5. Conformity Assessment | Select the appropriate route based on device class. May involve Notified Body. | Self-declaration (low risk) vs. Notified Body review (higher risk). | Manufacturer and Notified Body (if applicable) |
| 6. Notified Body Audit (if required) | Assessment of QMS and/or Technical Documentation by a designated third party. | Rigorous review of processes and data. Requires thorough preparation. | Notified Body |
| 7. EU Authorized Rep (if non-EU) | Appoint a legal entity within the EU to represent your company. | Verification of conformity, point of contact for authorities. Needed for market access. | Manufacturer & Appointed Representative |
| 8. Declaration of Conformity | Manufacturer issues a formal document confirming compliance with EU regulations. | Legally binding statement based on successful assessment. | Manufacturer |
| 9. Affix CE Mark | Apply the official CE mark to the device and associated documentation. | Visible, legible mark signifies compliance. | Manufacturer |
| 10. Post-Market Surveillance | Ongoing monitoring of device performance and safety in the market. | Includes vigilance reporting, complaint handling, PMS reports. Continuous obligation. | Manufacturer |
Frequently Asked Questions
Which countries accept the CE mark for medical devices?
Pretty much all of Europe, mate. It’s the passport for medical devices across the European Economic Area (EEA), which includes the EU countries plus Iceland, Liechtenstein, and Norway. Switzerland too, through special agreements. So, if you want to sell your medical gizmos there, you need that CE certification.
Is CE required in Europe?
Yeah, absolutely. If your medical device is going to be sold anywhere in the European Union or the EEA, you’ve gotta have the CE mark. It’s not optional, it’s a legal requirement that shows your product meets all the Needed safety and health standards. No CE marking, no sale.
What is the European CE approval?
It’s basically the stamp of approval that says your medical device complies with all the relevant European Union directives and regulations. Think of it as a declaration by you, the manufacturer, that your product is safe and effective for its intended use. It involves rigorous testing and documentation to prove you’ve met all the EU health and safety standards.
What is the EU certificate for medical devices?
This usually refers to the Certificate of Conformity or a similar document issued by a Notified Body. It’s the proof that an independent third party has assessed your medical device and confirmed it meets the requirements of the applicable EU regulations, like the Medical Device Regulation (MDR). It’s a key piece of the puzzle for obtaining your CE certification.
What’s the difference between MDR and IVDR for CE marking?
Because they cover different types of devices, that’s the main thing. The MDR (Medical Device Regulation) is for general medical devices – things like surgical tools, implants, and bandages. The IVDR (In Vitro Diagnostic Regulation) is specifically for devices used to test samples from the human body, like blood glucose meters or COVID-19 tests. Both require CE marking, but the rules and requirements are tailored to the specific risks associated with each type of device.